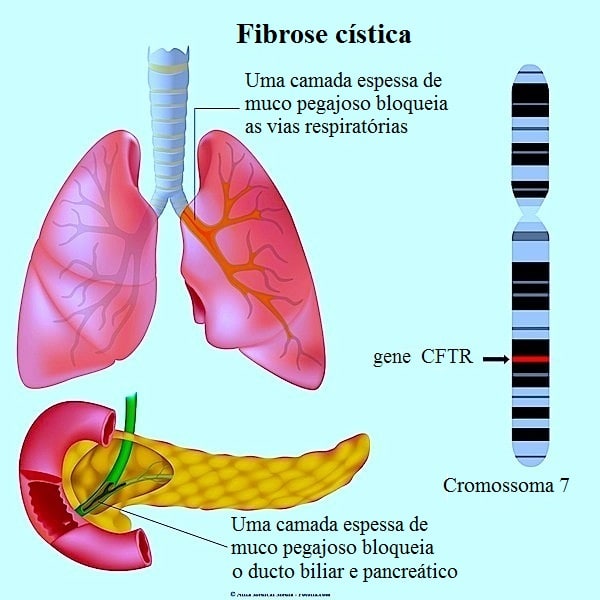
A fibrose cística é uma doença que afeta as glândulas exócrinas (que liberam as substâncias produzidas fora do corpo ou em órgãos que se comunicam com o exterior).
| ÍNDICE |
Diagnóstico da fibrose cística
A fibrose cística pode ser diagnosticada em três fases:
- Triagem pré-natal,
- Fase pós-natal,
- Fase precoce.
O diagnóstico precoce pode prevenir os danos pulmonares que já começam na infância, como as bronquiectasias.
Triagem pré-natal
Nas mulheres grávidas, podem ser feitos dois exames chamados de:
- Biópsia das vilosidades coriônicas que é uma pequena coleta de vilosidades corais (parte da placenta),
- Amniocentese: coleta de líquido amniótico.
Nessas amostras são examinados:
- o DNA das células para procurar possíveis mutações genéticas no feto,
- As enzimas intestinais fetais. Os níveis das enzimas intestinais fetais são reduzidos em um feto com fibrose cística.
Após o parto
Nos recém-nascidos, é efetuado o teste do tripsinogênio imunorreativo (IRT).
Os níveis elevados de tripsinogênio indicam que a criança tem fibrose cística.
Para confirmar o diagnóstico, os médicos podem também efetuar:
- Exame da função pulmonar,
- Cultura do escarro,
- Exames das fezes,
- Radiografia do tórax,
- Os exames genéticos são usados para detectar o gene da fibrose cística. Esses exames são indicados para os casais de portadores saudáveis com alto risco de terem filhos doentes.
Nesses casos, os testes fornecem resultados confiáveis.
Em outros casos, esses exames podem dar resultados errados e a doença não é diagnosticada.
Fibrose cística
© fotolia.com
Teste de suor para fibrose cística
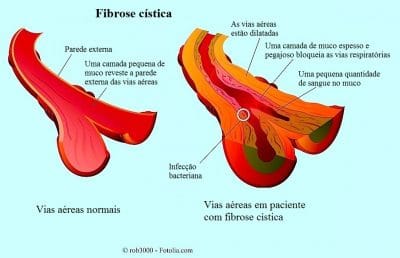
A função das glândulas sudoríparas inclui a secreção e a reabsorção de cloreto de sódio e de outros sais minerais.
As pessoas com fibrose cística têm as glândulas sudoríparas que não absorvem corretamente o sódio e o cloro.
A consequência é uma alta concentração de cloro e de sódio no suor.
Para o diagnóstico da fibrose cística, o teste do suor é o exame principal.
O teste do suor (para a coleta e dosagem dos eletrólitos no suor) é utilizado para o diagnóstico de fibrose cística:
- Em crianças,
- Nos adolescentes.
Hoje em dia, este teste faz parte do programa de triagem neonatal em muitos países.
Valores de cloro no suor
Adultos e crianças:
- Se a concentração de cloro for de pelo menos 60 mmol/L, o resultado é positivo.
- Se o valor for inferior a 40 mmol/L, o resultado é negativo (pessoa saudável).
- O intervalo entre 40 e 60 mmol/L é considerado borderline (limítrofe, é uma forma leve da fibrose cística). Nesse caso, aconselha-se repetir o exame.
Recém-nascidos (0-6 meses):
- Uma concentração de cloreto menor de 30 mmol/L é considerada normal.
- Se a concentração for maior do que 60 mmol/L, o recém-nascido sofre de fibrose cística.
- O nível limítrofe é de 30-60 mmol/L (fibrose cística atípica).
© fotolia.com
Tratamento para fibrose cística
Atualmente, não existe uma cura definitiva para esta doença.
Terapia genética
Alguns tratamentos foram desenvolvidos para tratar a mutação genética da fibrose cística.
O tratamento consiste em inserir uma cópia do gene saudável nas células do tecido pulmonar através de:
- Partículas virais,
- Aerossol.
Desta forma, as células deveriam produzir corretamente a proteína CFTR porque copiam a informação do gene saudável.
Atualmente, os resultados desta terapia são escassos, mas alguns estudos experimentais estão em andamento.
Tratamento para os pulmões
As infecções respiratórias são frequentes em pessoas com fibrose cística.
- Os antibióticos (orais, inalados ou por via intravenosa) são administrados para tratar as infecções.
A azitromicina (Zitromax) é a mais prescrita porque os estudos científicos têm mostrado melhorias em pacientes com fibrose cística, especialmente para tratar a bactéria Mycobacterium abscessus que é muito resistente aos outros medicamentos. - Os medicamentos anti-inflamatórios, como ibuprofeno, podem ser prescritos para reduzir a inflamação nos pulmões.
- Os broncodilatadores, tais como salbutamol (Aerojet) são recomendados pelo médico para relaxar os músculos da respiração.
- A solução salina hipertônica e acetilcisteína são prescritas para fluidificar o muco (diminuem a viscosidade).
- Para aumentar o nível de oxigênio no sangue, pode ser efetuada a oxigenoterapia.
Se o paciente sofrer de complicações pulmonares graves, o médico pode recomendar o transplante de pulmão (ambos os pulmões).
Tratamento para o sistema digestivo
Os problemas digestivos podem ser reduzidos com uma dieta bem equilibrada.
As enzimas pancreáticas por via oral são prescritas para:
- Facilitar a digestão das proteínas e das gorduras,
- A absorção de vitaminas.
Os suplementos de vitamina A, D, E e K são recomendados para aumentar o nível de vitaminas lipossolúveis.
Outros tratamentos para problemas digestivos são:
- Enemas, para facilitar a evacuação,
- Os medicamentos para fluidificar o muco, para evitar a obstrução do intestino (obstrução intestinal),
- Os medicamentos antiácidos para aliviar a azia e aumentar os efeitos terapêuticos das enzimas pancreáticas orais.
Fisioterapia para a fibrose cística
Para a drenagem das secreções, a fisioterapia respiratória pode ser muito útil.
O objetivo é estimular e facilitar a tosse que é o mecanismo para expulsar o muco do corpo.
Existem algumas terapias que precisam ser feitas com o fisioterapeuta, por exemplo, vibrações em posições diferentes.
Além disso, alguns exercícios com a garrafa PEP ou outras ferramentas devem ser feitos em casa regularmente.
A atividade esportiva regular faz parte da terapia.
Para ter bons resultados, o paciente deve dedicar cerca de 2/3 horas por dia aos tratamentos:
- Aerossol,
- Fisioterapia respiratória,
- Atividades esportivas.
Expectativa de vida e prognóstico do paciente com fibrose cística
Embora ainda não exista uma tratamento para fibrose cística, muitos anos de pesquisa produziram alguns resultados positivos.
O mais importante é que a expectativa de vida melhorou, embora a cura não seja possível.
Agora, a idade média de sobrevivência é de 35 a 40 anos devido aos avanços no diagnóstico e tratamento.
Antes de 1950, as crianças nascidas com esta doença sobreviviam menos de 6 anos.
Os pacientes querem saber o tempo de vida; não é fácil dar uma resposta a esta pergunta porque depende de muitos elementos.
Os fatores que podem afetar a expectativa de vida de pacientes com fibrose cística estão listados abaixo:
- Função do pâncreas: um funcionamento normal do pâncreas é um fator vital que desempenha um papel importante para melhorar a taxa de sobrevivência.
Os recém-nascidos que sofrem de fibrose cística atípica podem viver mais de 50 anos se a função pancreática for boa. - Idade: a expectativa de vida de uma pessoa nascida na década de 1980 é menor do que aquelas nascidas na década de 1990 ou depois.
As pessoas nascidas recentemente têm melhores opções de tratamento. - Outras doenças: a infecção crônica por Pseudomonas aeruginosa pode agravar os sintomas da fibrose cística e afeta negativamente a duração da vida.
Embora a alergia tenha um efeito negativo, existe apenas uma diferença marginal na sobrevivência dos pacientes. - Tipo de mutação: a gravidade dos sintomas e as taxas de sobrevivência também podem variar em função do tipo de mutação genética.
A fibrose cística é causada pela alteração do gene CFTR.
As mutações menos frequentes podem causar sintomas mais leves e permitem uma maior sobrevivência.
As estatísticas mostram que a expectativa de vida é maior em pacientes que realizaram o transplante de pulmão.
Hoje em dia, os programas de triagem neonatal em muitos países incluem exames para a fibrose cística, portanto o tratamento pode começar logo que possível.
Como prevenir a fibrose cística?
Podem ser feitos exames genéticos em membros da família de uma pessoa que sofre de fibrose cística.
Isso serve para descobrir se ela pode ter o gene responsável pela doença.
Um casal que pretenda ter um filho deve fazer a análise de DNA para:
- Reconhecer quaisquer mutações genéticas,
- Reduzir o risco de transmissão de doenças genéticas para o bebê.
Dieta e alimentação para a fibrose cística
De acordo com a medicina convencional
Uma pessoa que sofre de fibrose cística precisa de uma dieta com alto teor de calorias e proteínas, cerca de 50% a mais do que as pessoas saudáveis.
A razão é a má absorção, o indivíduo doente não consegue absorver os nutrientes dos alimentos para as necessidades do organismo.
Além disso, uma pessoa que tem uma alimentação saudável é capaz de lutar contra:
- Infecções pulmonares,
- Outras doenças.
É importante uma dieta rica em alimentos gordurosos porque quem sofre de fibrose cística perde muita gordura com as fezes.
Dado que muitas pessoas com fibrose cística sofrem de diabetes, é preciso evitar:
- Doces,
- Cereais.
De acordo com o higienismo (um tipo de medicina natural)
Os higienistas não tratam o sintoma, mas aconselham um tipo de alimentação e um estilo de vida para prevenir a doença.
No caso da fibrose cística, a dieta hiperproteica sobrecarrega o trato gastrointestinal e especialmente o pâncreas, que não funciona adequadamente por causa da doença.
De acordo com o higienismo, o alimento principal é a fruta porque:
- Contém aminoácidos em vez de proteínas. As proteínas são estruturas muito complexas, são constituídas por muitos aminoácidos unidos entre si. Para o corpo, a digestão dos aminoácidos é mais fácil e rápida porque não precisa quebrar as ligações químicas entre os aminoácidos. Cerca de 1-2% do peso das frutas e vegetais é constituído por aminoácidos.
- Contém vitaminas e minerais
- Digere-se rapidamente.
Para reduzir a produção de muco e prevenir as infecções, os higienistas recomendam uma dieta à base de:
- Frutas,
- Vegetais crus,
- Batatas,
- Sementes e frutas com casca (sem exagerar).