La talasemia es una enfermedad hereditaria causada por un defecto en la producción de la hemoglobina.
La hemoglobina es un pigmento que se encuentra en los glóbulos rojos que se une con:
- Oxígeno
- Dióxido de carbono
Esta unión permite que los glóbulos rojos puedan transportar el oxígeno a las células y el dióxido de carbono a los pulmones.
La hemoglobina tiene 2 cadenas alfa y 2 cadenas beta que se unen entre ellas.
Es importante que la cantidad de cadenas de la hemoglobina alfa y beta sea igual en los glóbulos rojos (eritrocitos) para garantizar la supervivencia y un funcionamiento adecuado de estas células.
La falta de las cadenas de hemoglobina alfa provoca la acumulación de hemoglobina beta en los glóbulos rojos y viceversa.
Por consiguiente, las cadenas libres precipitan y pueden afectar a los eritrocitos.
Estos grupos de cadenas se oxidan y pueden provocar:
- Hemolisis (muerte de los glóbulos rojos) debido al deterioro de la membrana celular
- Eritropoyesis (proceso de formación de los glóbulos rojos) ineficaz
La gravedad de esta enfermedad depende del desequilibrio de la cantidad de las cadenas de globina.
Evolución y consecuencias de la talasemia
La disminución de glóbulos rojos provoca anemia por lo que el organismo intenta compensar esta situación aumentando la absorción de hierro en los alimentos.
El bazo filtra los eritrocitos deteriorados y después los destruye.
Los efectos de la talasemia en el bazo son:
- Sobrecarga
- Esplenomegalia (agrandamiento del bazo)
Con el tiempo, el bazo agrandado también retiene los glóbulos blancos y las plaquetas.
Esto puede causar:
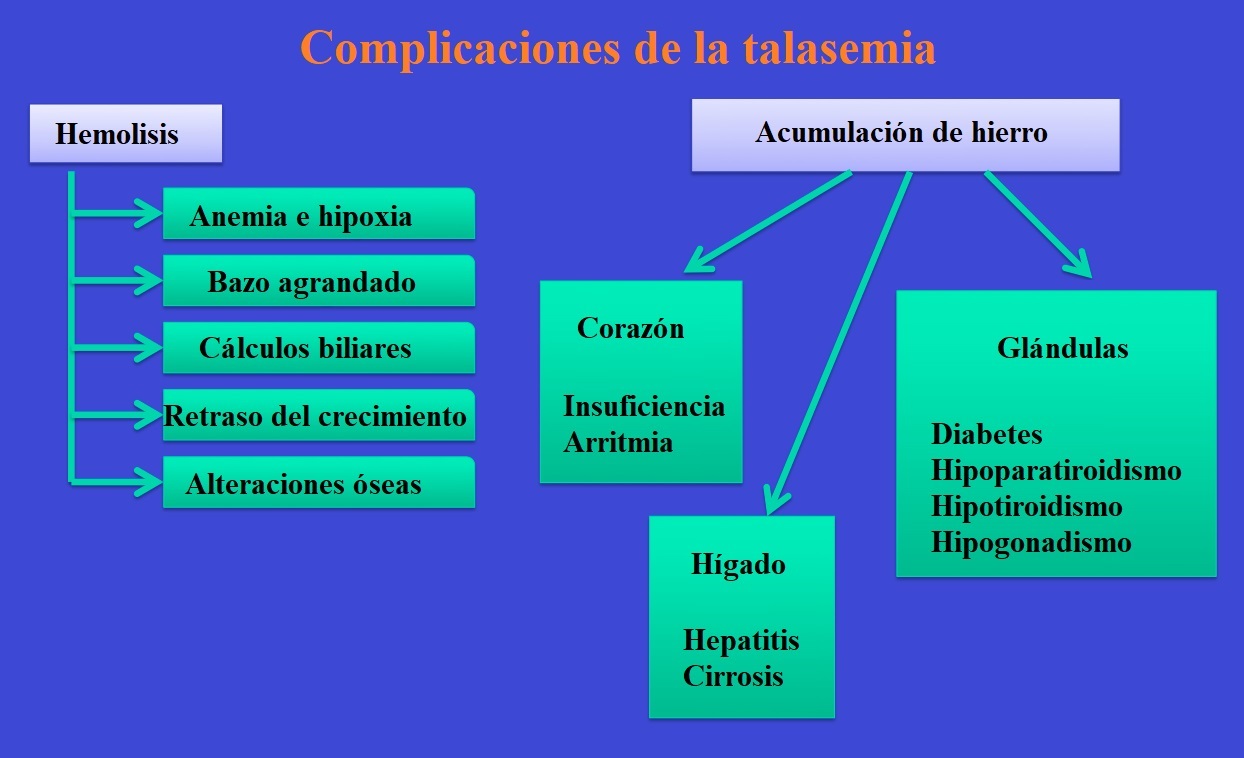
Entre las complicaciones de la talasemia se encuentran:
- Anoxia (falta de oxígeno en las células)
- Retraso del crecimiento en los niños y retraso del desarrollo puberal
- Cálculos biliares, la producción excesiva de bilirrubina debido a la hemolisis crónica provoca a formación de cálculos biliares pigmentados (EVERSON G. T.: Gallbladder function in gallstone disease. Clin. North Am. 20 (1991), 85)
- Enfermedades de los huesos y osteoporosis por el agrandamiento y la hiperactividad de la médula ósea (dentro de los huesos)
La dilatación de la médula ósea provoca el deterioro de la capa cortical del hueso y esto puede provocar la formación de hueso nuevo: cráneo «en cepillo».
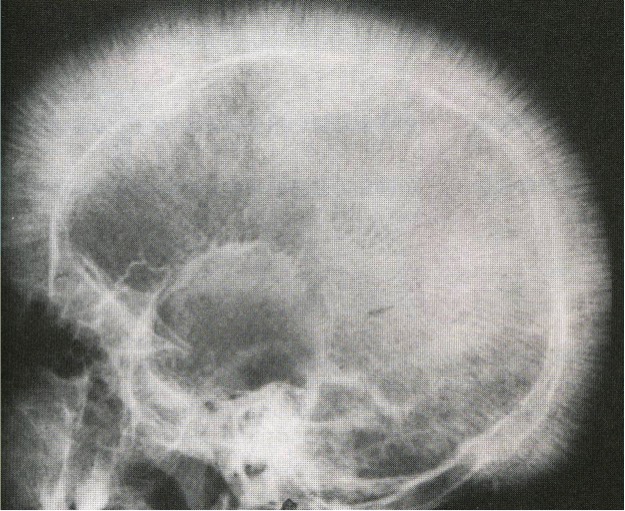
© Massimo Defilippo
Por esta razón, las personas afectadas requieren trasfusiones de sangre y esto provoca una acumulación de hierro.
El hierro es necesario para el organismo, pero un átomo libre de hierro provoca la formación de radicales libres que son tóxicos para el organismo.
En condiciones normales, el hierro siempre se une a otras proteínas: ferritina, transferritina, etc.
Cuando estas proteínas están saturadas de hierro, este metal se acumula en los siguientes órganos:
- Hígado (primer órgano afectado), puede provocar hepatitis crónica y cirrosis
- Corazón (cuando el hígado ya está saturado), provoca arritmia e insuficiencia cardíaca
- Glándulas, puede provocar: hipogonadismo, hipotiroidismo, diabetes, hipoparatiroidismo
Fuente: Borgna-Pignatti C (Department of Clinical and Experimental Medicine, University of Ferrara and Division of Pediatrics, Arcispedale Sant’Anna, Via Savonarola 9, 44100, Ferrara, Italy)
Esto puede llegar a causar la muerte de las personas, esto ocurre debido a la cardiopatía y no debido a una hepatopatía, ya que una pequeña cantidad de hierro es suficiente para alterar el sistema de conducción eléctrica del corazón.
© Massimo Defilippo
¿Cómo se transmite la talasemia?
La causa de la talasemia siempre es genética: es una enfermedad autosómica recesiva. Uno o ambos padres son portadores sanos de la enfermedad.
Pueden transmitirles los genes a sus hijos.
Clasificación de la talasemia
Existen dos tipos de cadenas globínicas: las alfa y las beta.
| Alfa |
|
| Beta |
|
Talasemia alfa
La talasemia alfa provoca un exceso de globina beta, que a su vez causa la formación de tetrámeros de cadenas de globina beta (β4), esto se conoce como enfermedad de la hemoglobina H.
Los tetrámeros β4 son solubles, pero en caso de oxidación pueden provocar hemolisis.
La enfermedad de la hemoglobina H Constant Spring es una forma grave de esta alteración hemolítica.
La talasemia más grave es la talasemia alfa mayor en la que el feto no produce cadenas de globina alfa, esta situación no es compatible con la vida.
Los genes de la alfa globina se encuentran en el cromosoma 16.
La cadena de hemoglobina alfa está formada por 4 unidades globulares.
Cada una de ellas está codificada por un gen distinto en el cromosoma.
Los síntomas y las complicaciones de la enfermedad dependen de la cantidad de genes ausentes y mutados.
- Los portadores sanos no tienen signos de esta enfermedad.
En caso de 2 genes ausentes o mutados, el paciente desarrolla talasemia alfa o talasemia menor. Esta enfermedad causa una anemia leve. - En caso de 3 genes mutados o ausentes, el paciente desarrolla la enfermedad de la hemoglobina H (se diagnostica a través de un análisis de sangre).
Esta forma de talasemia causa una anemia moderada o grave. - Si todos los genes están ausentes o si han mutado, la talasemia es muy grave y el bebé nace muerto o muere poco después del parto.
Un niño hereda cuatro genes de alfa globina (dos de cada padre).
El hijo de una pareja afectada por rasgo talasémico (cada uno de los padres con 2 genes ausentes o mutados) tiene:
- 25% de probabilidad de estar enfermo (3 o 4 genes anormales)
- 50% de probabilidades de ser un portador sano
- 25% de ser sano con 1 sólo gen anormal
Si al padre le faltan dos genes de globina alfa y a la madre le falta un gen de globina alfa, cada niño tiene una probabilidad del 25% de heredar:
- Cuatro genes normales (ninguna anemia)
- Un gen ausente y tres genes normales (portador sano)
- Dos genes ausentes y dos genes normales (enfermedad)
- Tres genes ausentes y un gen normal (hemoglobinapatía H)
La mayoría de las personas con talasemia alfa tienen síntomas leves de la enfermedad.
Talasemia beta
La talasemia beta provoca un exceso de globina alfa que forman tetrámeros de globina alfa (α4) en lugar de dos cadenas alfa y dos pares de cadenas alfa y dos pares de cadenas beta.
Estas formaciones no son solubles y se acumulan en los eritroblastos (células precursoras de los glóbulos rojos) y afectan a:
- Eritropoyesis
- Proceso de maduración de las células
- Membrana celular
Esto provoca anemia y la formación de glóbulos rojos que no funcionan de manera adecuada.
Las cadenas de globulina beta están codificadas por dos genes distintos.
En caso de ausencia o mutación:
- De un sólo gen, la anemia es leve o ausente
- De ambos genes, los síntomas son graves
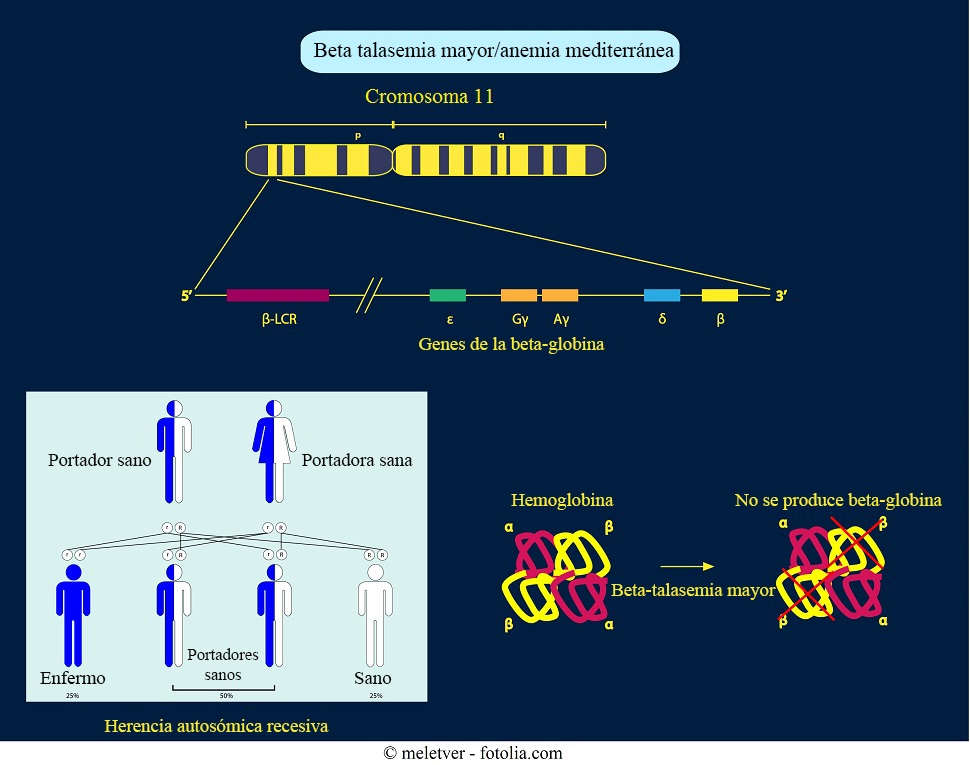
El gen beta globina se encuentra en el cromosoma 11.
Un niño hereda dos genes beta globina (uno de cada padre). Si estos genes están ausentes o alterados se produce la talasemia beta. Esto significa que el organismo no produce suficientes cadenas de hemoglobina beta.
- Las personas con un solo gen alterado son portadores sanos porque se trata de un gen recesivo. Esta enfermedad se llama talasemia beta menor y causa una anemia moderada.
- Si ambos genes están alterados o ausentes, se produce la talasemia beta intermedia o la talasemia beta mayor (también llamada anemia de Cooley).
© fotolia.com
La forma intermedia causa una anemia moderada. La forma mayor causa anemia grave.
Si cada uno de los padres tiene un gen alterado o ausente cada niño tiene:
- El 25% de probabilidad de heredar dos genes normales (ninguna anemia)
- El 50% de probabilidad de heredar un gen alterado y un gen normal (talasemia beta menor). En este caso, el paciente puede tener anemia leve o, en caso de talasemia mínima, no presenta ningún síntoma
- El 25% de probabilidad de heredar dos genes alterados (talasemia beta mayor).
¿Cuál es la diferencia entre talasemia mayor y menor?
Hay dos formas de talasemia beta: la talasemia menor (o benigna) y la talasemia mayor (también conocida como anemia de Cooley).
La talasemia menor: la persona tiene solo un gen ausente o alterado (junto al gen de una cadena beta normal). El sujeto se denomina heterocigoto para talasemia beta.
Las personas con talasemia menor tienen una anemia ligera con una leve reducción del nivel de hemoglobina en la sangre.
Las personas con talasemia menor tienen un nivel de hierro normal en la sangre. No es necesario ningún tratamiento para la talasemia menor.
La talasemia mayor (o anemia de Cooley): los individuos con talasemia mayor tienen dos genes mutados a causa de la “talasemia beta “y ningún gen beta normal.
El sujeto es homocigoto para talasemia beta.
Esto causa una carencia de la producción cadena beta.
La talasemia mayor es una enfermedad grave.
El cuadro clínico de la talasemia mayor fue descrito en 1925 por el pediatra estadounidense Thomas Cooley.
El niño con talasemia mayor parece sano al nacer.
La hemoglobina predominante al nacer es la hemoglobina fetal (HbF). La HbF tiene dos cadenas alfa (como AHb) y dos cadenas gamma (a diferencia de Hb A).
Al no tener ninguna cadena beta, al nacer el niño está protegido por los efectos de la talasemia mayor.
El favismo es una enfermedad causada por una deficiencia enzimática: la glucosa-6-fosfato deshidrogenasa -G-6-PDH. Las personas que sufren de esta enfermedad pueden tener anemia con ictericia por la destrucción de los glóbulos rojos cuando comen habas, guisantes y otras hortalizas.
También puede aparecer después de haber tomado algunos medicamentos como salicilatos y sulfamidas.
Talasemia durante el embarazo
Por lo general, el embarazo suele ser seguro en las mujeres con talasemia beta.
Hoy en día, las mujeres con talasemia mayor intermedia pueden tener hijos. (Thalassaemia in pregnancy.Leung TY – Lao TT Best Pract Res Clin Obstet Gynaecol. 2012 Feb; 26(1):37-51.).
Los factores más importantes que hay que valorar a través de una resonancia magnética, son la funcionalidad cardíaca y la cantidad de hierro.
© fotolia.com
Síntomas de la talasemia
Los síntomas de la talasemia beta dependen de la gravedad de la enfermedad.
Los portadores sanos no muestran ningún síntoma.
Los individuos afectados por talasemia grave pueden mostrar los siguientes síntomas desde los primeros meses de vida:
- Anemia grave,
- Cansancio y debilidad
- Falta de aliento
- Crecimiento lento
- Palidez en la cara (uno de los primeros síntomas que se observan en los bebés)
- Orina de color oscuro
- Hinchazón del hígado y del bazo
- Ictericia
Las personas que practican deporte y tienen un valor de hemoglobina inferior a 9 g/ dl y un rendimiento físico considerablemente más bajo.
Entre las consecuencias de la talasemia está prohibido donar sangre si los valores de hemoglobina son inferiores a:
- 13.5 g/dl en los hombres
- 12.5 g/dl en las mujeres
- Talasemia: diagnóstico, alimentación y pronóstico
- Anemia por deficiencia de hierro: causas y síntomas
- Pólipos intestinales: causas, clasificación y síntomas