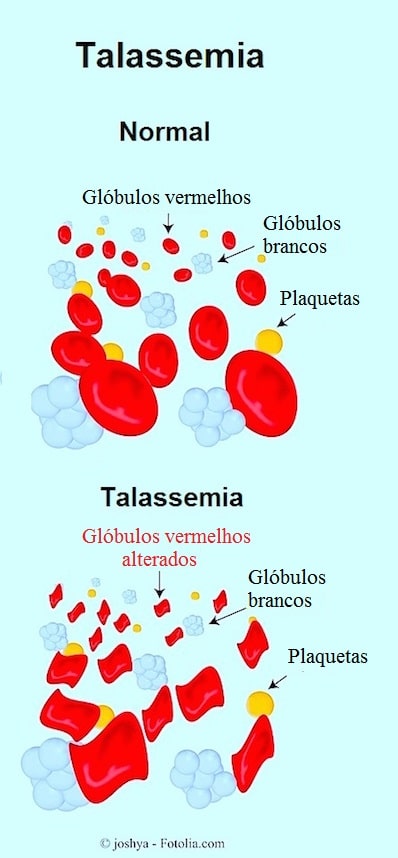
A talassemia é uma doença hereditária causada por um defeito na produção de hemoglobina.
| INDICE |
A hemoglobina é um pigmento contido nos glóbulos vermelhos que se liga:
- Ao oxigênio,
- Ao dióxido de carbono.
Esta ligação permite que os glóbulos vermelhos transportem o oxigênio para as células e o dióxido de carbono para os pulmões.
A hemoglobina contém de 2 cadeias alfa e 2 cadeias beta ligadas.
É essencial que as quantidades de cadeias de globina alfa e beta sejam iguais nos glóbulos vermelhos (eritrócitos) para o bom funcionamento e a sobrevivência dessas células.

© fotolia.com
A falta de cadeias de hemoglobina alfa causa uma acumulação de hemoglobina beta nos glóbulos vermelhos e vice-versa.
A conseqüência é a formação de precipitados (grupos de cadeias) que podem prejudicar os eritrócitos.
Esses agregados se oxidam e podem causar:
- Hemólise (morte de células vermelhas) porque danificam a membrana celular,
- Eritropoiese (processo de formação de glóbulos vermelhos) ineficaz.
A gravidade da doença depende do desequilíbrio na quantidade das cadeias globinas.
Curso e consequências da talassemia
A diminuição dos glóbulos vermelhos causa anemia e o corpo tenta compensar isso aumentando a absorção de ferro nos alimentos.
O baço filtra os eritrócitos danificados e os destrói.
As conseqüências da talassemia no baço são:
- Sobrecarga,
- Esplenomegalia (aumento do tamanho do órgão).
Ao longo do tempo, o baço aumentado também mantém os glóbulos brancos e as plaquetas dentro.
As conseqüências podem ser:
Entre as complicações da talassemia existem:
- Anoxia (falta de oxigênio nas células),
- Retardo no crescimento em crianças e desenvolvimento tardio da puberdade.
- Os cálculos biliares, a produção excessiva de bilirrubina devido à hemólise crônica é um pré-requisito para a formação de cálculos cíclicos pigmentados (EVERSON G. T.: Gallbladder function in gallstone disease. Gastroenterol. Clin. North Am. 20 (1991), 85).
- Alterações ósseas e osteoporose devido à ampliação e hiperatividade da medula óssea (é encontrada dentro dos ossos),
- A expansão da medula óssea provoca o desgaste a parte cortical do osso, entre as conseqüências existe uma nova formação óssea com aspecto radiográfico de “cabelos em pé”.
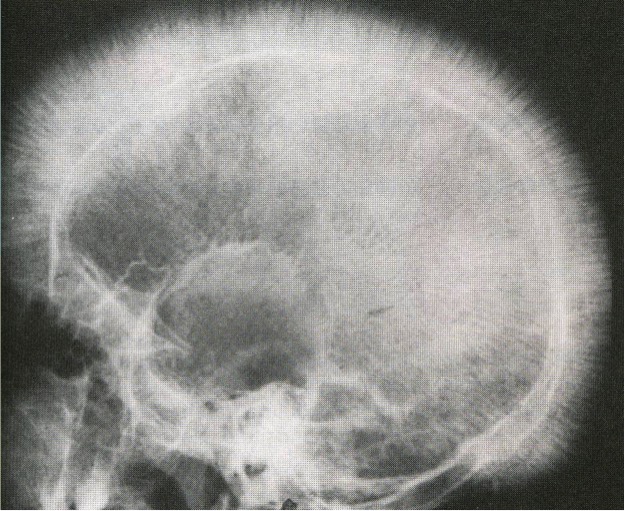
Radiografia com aspecto de “cabelos em pé”
© Massimo Defilippo
A necessidade de transfusões de sangue causa um acúmulo de ferro.
O ferro é necessário no corpo, mas um átomo de ferro livre causa a formação de radicais livres que são tóxicos para o corpo.
No corpo, o ferro sempre se liga às proteínas: ferritina, transferrina, etc.
Quando estas proteínas são saturadas de ferro, este metal se acumula nos órgãos:
- O fígado (é atingido primeiro), pode causar hepatite crônica e cirrose,
- O coração (quando o fígado já está saturado) causa arritmia e insuficiência cardíaca,
- As glândulas, as conseqüências podem ser: hipogonadismo, hipotireoidismo, diabetes, hipoparatiroidismo, Fonte: Borgna-Pignatti C – (Department of Clinical and Experimental Medicine, University of Ferrara and Division of Pediatrics, Arcispedale Sant’Anna, Via Savonarola 9, 44100, Ferrara, Italy)
A morte ocorre devido a doenças cardíacas e não por causa de doenças hepáticas porque uma pequena quantidade de ferro é suficiente para alterar a condução nervosa do coração.

© Massimo Defilippo
Transmissão da talassemia
A causa da talassemia é genética: é uma doença autossômica recessiva. Um ou ambos os pais são portadores da doença.
Os genes podem passar para suas crianças.
Classificação da talassemia
Existem dois tipos de cadeias de síntese de globina: alfa e beta.
| Alfa |
|
| Beta |
|
Alfa talassemia
A talassemia alfa causa um excesso de beta-globina, o que leva à formação de tetrâmeros de beta globina (β4), esta doença é chamada de doença de hemoglobina H.
Os tetrâmeros β4 são solúveis, mas em caso de oxidação podem causar hemólise.
A Hemoglobina Constant Spring é uma forma grave deste transtorno hemolítico.
A talassemia mais séria é a talassemia alfa Maior, na qual o feto não produz alfa-globina, esta situação é incompatível com a vida.
Os genes da Alfa-globina estão localizados no cromossomo 16.
A cadeia alfa da hemoglobina é constituída por 4 subunidades globulares.
Cada subunidade é codificada por um gene diferente no cromossomo.
Os sintomas e as complicações da doença dependem do número de genes ausentes ou mutados:
- Com dois genes anômalos (ou ausentes), ocorre a talassemia tipo alfa ou talassemia menor. Esta patologia causa uma anemia leve.
- No caso de 3 genes mutantes ou ausentes, o paciente desenvolve a doença de hemoglobina H (diagnosticada com exames de sangue).
Esta forma de talassemia provoca uma anemia moderada ou grave. - Se todos os genes estão ausentes ou mutados, a talassemia é muito séria e o bebê nasce morto ou a morte ocorre logo após o nascimento.
Uma criança herda quatro genes de alfa-globina (dois de cada pais).
O filho de dois pessoas que sofrem de traço talassêmico (ambos com 2 genes ausentes ou mutados) tem:
- 25% de chance de ser doentes (3 ou 4 genes anômalos),
- 50% de chance de ser portadores,
- 25% de ser saudáveis ou com 1 gene anormal.
Se dois genes de Globina faltam ao pai e a mãe não possui um gene de Globina alfa, cada criança tem um 25% de chance de herdar:
- Quatro genes normais (sem anemia),
- Um gene ausente e três genes normais (portador saudável).
- Dois genes “ausentes” e dois genes normais (doença),
- Três genes faltantes e um gene normal (Hemoglobinopatia H),
A maioria das pessoas com talassemia alfa tem os sintomas mais leves da doença.
Talassemia beta
A talassemia beta causa um excesso de alfa globinas que formam tetrâmeros (α4) em vez de dois pares de cadeias alfa e dois pares de cadeias beta.
Essas formações insolúveis se acumulam no eritroblasto (glóbulo vermelho imaturo) e interferem com:
- Eritropoiese,
- Maturação celular,
- Membrana celular.
A consequência é a anemia e a formação de glóbulos vermelhos que não funcionam.
As cadeias beta são codificadas por dois genes diferentes.
Em caso de falta ou mutação:
- De um único gene, a anemia é leve ou ausente,
- De dois genes, os sintomas são sérios.
O gene da beta-globina está localizado no cromossomo 11.
Uma criança herda dois genes beta-globina (um de cada pai).
Se estes genes faltam ou são alterados ocorre a beta-Talassemia. Isto significa que o corpo não produz suficiente cadeias beta da hemoglobina.
- Quem possui apenas um gene alterado é um portador saudável porque é um gene recessivo. Esta patologia é chamada beta Talassemia minor e provoca uma leve anemia.
- Se ambos os genes faltam ou são alterados, ocorre a beta-talassemia intermediária ou beta talassemia major (também conhecida como anemia de Cooley).

© fotolia.com
A forma intermediária provoca uma leve anemia. A forma maior causa uma anemia grave.
Se cada pai tem um gene alterado, cada criança tem:
- Uma probabilidade de 25% de herdar dois genes normais (sem anemia),
- Uma probabilidade do 50% de herdar um gene alterado e um gene normal (beta-Talassemia minor). Neste caso, o paciente pode ter anemia leve ou em caso de talassemia mínima não apresenta sintomas.
- 25% de chance de herdar dois genes alterados (beta-talassemia major).
Diferença entre talassemia major e minor
Existem duas formas de beta-Talassemia: talassemia minor (ou benigna) e a talassemia major (também conhecida como anemia de Cooley).
A talassemia minor: a pessoa tem um gene alterado ou ausente (junto com o gene de uma cadeia beta normal). O indivíduo se diz heterozigoto para a beta-Talassemia.
Pessoas com talassemia minor tem uma leve anemia com leve redução do nível de hemoglobina no sangue.
As pessoas com talassemia minor, tem um nível normal de ferro no sangue. Não é necessário nenhum tratamento para a talassemia minor.
A talassemia major: os indivíduos com talassemia major possuem dois genes mutantes para a “beta-Talassemia” e nenhum gene beta normal.
O indivíduo é homozigoto para a beta-Talassemia.
Isso resulta em uma carência na produção cadeia beta.
A talassemia major é uma doença grave.
O quadro clínico da talassemia major foi descrito em 1925 pelo pediatra americano Thomas Cooley.
A criança com talassemia major parecem saudavél ao nascer.
A hemoglobina predominante no nascimento é ainda a hemoglobina fetal (HbF). A HbF tem duas cadeias alfa (como A Hb) e duas cadeias gama (ao contrário da Hb A).
Não tendo nenhuma cadeia beta o bebê é protegido no nascimento pelos efeitos da talassemia major.
O favismo é uma doença causada pela deficiência de uma enzima: glicose-6-fosfato desidrogenase-G-6-PDH.
As pessoas que sofrem desta doença podem ter anemia com icterícia pela destruição das células vermelhas do sangue quando eles comem feijão, ervilhas e outros vegetais.
Pode ocorrer mesmo depois de tomar certos medicamentos, tais como salicílico e sulfonamidas.
Talassemia em gravidez
Geralmente, a gravidez é possível e segura em mulheres com β-talassemia.
Hoje, as mulheres com talassemia maior e talassemia intermediária podem ter filhos. (Thalassaemia in pregnancy – Leung TY – Lao TT Best Pract Res Clin Obstet Gynaecol. 2012 Feb; 26(1):37-51.).
Os fatores mais importantes para avaliar são a função cardíaca e o carregamento de ferro, usando a ressonância magnética.
© fotolia.com
Sintomas da talassemia
Os sintomas da talassemia beta dependem da gravidade da doença.
O portador não apresenta quaisquer sintomas.
O indivíduo que sofre de talassemia grave pode apresentar os seguintes sintomas desde os primeiros meses de vida:
- Anemia grave,
- Fadiga e fraqueza,
- Falta de ar,
- Atraso no crescimento,
- Palidez no rosto (um dos primeiros sintomas que observamos em crianças),
- Urina escura,
- Alargamento do fígado e do baço,
- Icterícia.
Aqueles que praticam esporte e tem um valor de hemoglobina inferior a 9 g/dl, tem uma diminuição no desempenho que pode ser importante.
Entre as consequências da anemia mediterrânea existe a proibição de doar sangue se os valores de hemoglobina são inferiores a:
- 13,5 g/dl em homens,
- 12,5 g/dl em mulheres.
Diagnóstico da talassemia
O médico diagnostica a talassemia moderada e grave na infância porque os sintomas aparecem nos primeiros dois anos de vida.
São feitos vários exames de sangue para o diagnóstico da talassemia:
Um exame de hemograma completo incluindo:
- Os valores de hemoglobina,
- O número e o tamanho das células vermelhas do sangue.
As pessoas com talassemia têm menos células vermelhas no sangue e menos hemoglobina do que o normal.
Os indivíduos com alfa/beta-talassemia têm células vermelhas menores.
Um adulto deve ter os valores de hemoglobina e hematócrito acima de:
1. Homem adulto -13 g/dl de hemoglobina e 39% de hematócrito;
2. Mulher adulta -12 g/dl de hemoglobina e 36% de hematócrito;
3. Mulher grávida -11 g/dl de hemoglobina e 33% de hematócrito.
A contagem dos reticulócitos (mede as células que dão origem a hemácia) pode indicar se a medula óssea não produz um número adequado de glóbus vermelhos.
As análises laboratoriais para o ferro indicam se a causa da anemia é:
- A deficiência de ferro,
- A talassemia.
Não devemos confundir os valores de ferritina com o ferro no sangue porque eles são dois dados diferentes.
Em indivíduos com anemia mediterrânea a ferritina pode ser normal.
A ressonância magnética é um exame útil porque mostra a quantidade de ferro acumulado no fígado e no coração
A bilirrubina indireta (não conjugada) é maior em pacientes com talassemia.
Os exames genéticos são usados para diagnosticar quando existe uma história familiar de Talassemia alfa.
Para diagnosticar a anemia mediterrânea pré-natal pode ser feita uma biópsia do vilo corial no feto logo nas 10° semana. Desta forma você pode saber se a criança tem talassemia.
© fotolia.com
Tratamento para talassemia
O tratamento padrão consiste em:
- Transfusões de sangue,
- Quelação do ferro.
Nos casos mais graves, é possível prosseguir com:
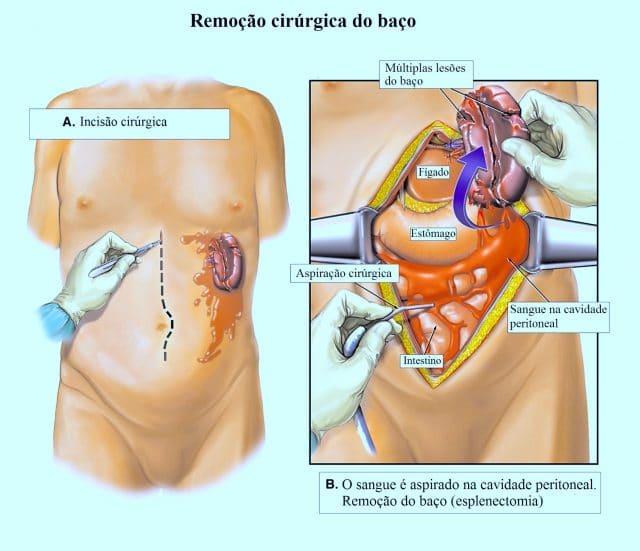
- Remoção do baço,
- Transplante da medula óssea.
A transfusão de sangue envolve a injeção de células vermelhas, através de uma veia para restaurar os níveis normais de glóbulos vermelhos e hemoglobina.
As transfusões são repetidas:
- A cada 4 meses para talassemia moderada,
- A cada 2-4 semanas para beta-talassemia major.
A quelação de ferro significa a remoção do excesso de ferro do corpo.
As transfusões de sangue podem causar sobrecarga de ferro e doenças cardíacas.
O médico deve administrar a terapia de quelação de ferro aos pacientes que recebem transfusões de sangue.
Os medicamentos usados para a quelação de ferro são:
- A deferoxamina, um líquido administrado por via subcutânea,
- O deferasirox, uma pílula que você toma por via oral.
© alamy.com
A esplenectomia (remoção do baço) pode ser necessária para os pacientes com doença de hemoglobina H.
O transplante de medula óssea é o tratamento mais eficaz. A compatibilidade entre dador e receptor ocorre quando o doador tem os mesmos tipos de proteínas (antígenos de leucócitos humanos HLA-) na superfície das células do receptor.
Um transplante de medula óssea de um irmão ou irmã oferece a melhor chance de cura.
A maioria dos pacientes com talassemia não possui um doador adequado.
As células-tronco da medula óssea transplantadas começam a gerar novas células sangüíneas.
Alimentação e dieta para talassemia
Em caso de anemia mediterrânea major é necessário tentar limitar os alimentos que contêm muito ferro.
A maior absorção de ferro no intestino é uma característica da talassemia.
Beber um copo de chá preto durante as refeições reduz a absorção de ferro dos alimentos, particularmente na talassemia intermediária (Alarcon, 1979).
No entanto, não há evidências de que uma dieta pobre em ferro seja útil na talassemia maior; Apenas os alimentos muito ricos em ferro devem ser evitados, tais como:
- O fígado,
- O chocolate,
- Leguminosas,
- Alguns coquetéis de vitaminas.
Cuidado com os alimentos para crianças, como cereais matinais e multivitaminicos, porque contêm ferro adicionado.
Cálcio
Muitos fatores na talassemia causam depleção de cálcio, então recomenda-se uma dieta com alimentos ricos neste mineral.
No entanto, para evitar a formação de cálculos, é melhor evitar os suplementos.
De acordo com a dieta do grupo sanguíneo, a talassemia desenvolveu-se principalmente nos países do mediterrâneo porque nesses estados a população sempre comiu muito glúten.
De acordo com esta teoria, na maioria dos casos, a eliminação de cereais contendo glúten pode reduzir os sintomas, portanto é recomendado evitar:
- Massa,
- Bolachas salgadas,
- Bolos,
- Pizza,
- Pão,
- Biscoitos,
- Espelta,
- Cevada,
- Aveia,
- Kamut,
- Sorgo.
Prognóstico dos pacientes com talassemia
Os pacientes com talassemia moderada têm uma boa chance de sobreviver, contanto que faça o tratamento necessário (transfusões e terapia de quelação de ferro).
A sobrecarga de ferro é a principal causa de morte em pacientes com talassemia, a terapia de quelação de ferro é extremamente importante.
O transplante de medula óssea pode curar a talassemia.
Prevenção
Os indivíduos portadores de talassemia beta devem ser informados sobre os riscos ligados à procriação com um parceiro com a mesma alteração genética.